Pharmacopeial identification and limit tests
Welcome to this page, where I want to share with you some interesting facts about how we check the quality of medicines. When we take medicines, we want to make sure they are safe, effective, and consistent. That’s why we use some tests to find out what is in the medicines and how much of it is there. These tests are called pharmacopeial identification (ID) and limit tests, and they are based on the rules made by some organizations working closely with government and regulatory authorities to ensure the safety of the medicines.
Let me explain what these tests are:
- Limit tests: are used to measure the amount of impurities and contaminants in the samples, such as heavy metals, arsenic, sulfates, chlorides, … These tests are required to ensure that the medicines are safe for human consumption by establishing a maximum limit level acceptable, hence the name, and it is not unusual to have concentrations in the range of parts per million (ppm).
- ID tests: These tests are used to confirm the identity and the ingredients of the medicines. They tell us if the medicines are what they claim to be.
There are different ways to do these tests, but here I focus on the ones that use chemistry. My goal is simple: to help you understand the basic science behind some of these tests. I will mainly talk about the tests that follow the rules of three major organizations:
- European Pharmacopoeia (EP)
- United States Pharmacopeia (USP)
- Japanese Pharmacopoeia (JP)
What You’ll Find Here
- Procedure: I will give you a brief overview of how each test is done and what kind of chemical reactions are involved.
- Science behind: I will explain to you the main ideas and concepts behind each test and why they work.
- References/sources: At the end of each section, you will find some links to the sources that I have used to get the information and to some more resources that you can check out if you want to learn more.
The Missing Pieces
Now, here’s where you come in! Some of the tests are still a mystery to me because I could not find enough reliable sources to explain them. If you know something that I don’t, please share it with me. I will be very grateful and I will mention your name on this page (unless you don’t want me to).
Please keep in mind that this is a work in progress: Right now, I only cover a few tests, but I will add more tests as I learn more about them.
Feel free to contact me if you have any questions or comments at the bottom of this page.
Outline
The outline for this page is:
Identity tests
Acetate
The following ID tests for acetates are used in the different general chapters:
- Based on the odour produced when acid is added.
- Based on the odour produced when acid and alcohol are added.
- Reaction with a solution of lanthanum nitrate.
- Reaction with iron (III) chloride.
Note: tests based on the smell (like 1 and 2 above) should be done with caution (or, preferably, not performed at all) as other components in the sample might produce harmful fumes.
Based on the on the odour when mixed with acid.
Reaction a of EP (2.3.1) and reaction 1 of JP <1.09>.
Procedure:
- Heat the sample with acid (oxalic acid in EP and dilute sulfuric acid in EP).
- The vapours produced have the characteristic odour of acetic acid and show an acid reaction (this last step only mentioned in EP).
- Reaction with a solution of lanthanum nitrate.
Science behind.
Since oxalic acid and sulfuric acids are stronger acids than acetic acid, an acetate salt will shift the equilibrium acetate (CH3-COO–)/acetic (CH3-COOH) towards the acetic acid, which is more volatile than sulfuric acid or oxalic acid, heating will produce vapours of acetic:
CH3-COO– + H+ ⇄ CH3-COOH↑
Sources:
- Ole Pederson. “Pharmaceutical Chemical Analysis”. (CRC press Taylor and Francis group. 2006).
Based on the on the odour when mixed with acid and alcohol.
Reaction 2 of JP <1.09>
Procedure:
- Heat the sample with acid sulfuric acid and a small quantity of ethanol.
- The vapours produced have the characteristic odour of ethyl acetate.
Science behind:
This is an esterification reaction, where the acetate (CH3-COO–) is transformed to acetic acid (CH3-COOH), see the previous reaction, and this will react with the ethanol (CH3-CH2OH) to form ethyl acetate (CH3-COO-CH2-CH3):
CH3-COO– + H+ → CH3-COOH
CH3-COOH + CH3-CH2OH → CH3-COO-CH2-CH3
Source:
Reaction with a solution of lanthanum nitrate.
This is reaction b of EP <2.3.1> and A of USP <191>, with the USP inserting additional steps.
Procedure as per EP
- Prepare an aqueous solution with the sample.
- Add a lanthanum nitrate solution.
- Add Iodine.
- Add diluted ammonia.
- Heat to boiling: a blue precipitate is formed or a dark blue colour developed.
Procedure as per USP (in red the additional steps from the EP).
- Prepare an aqueous solution with the sample.
- Adjust the pH of the solution with sodium hydroxide to be slightly alkaline.
- Add a lanthanum nitrate solution.
- If a white precipitate is formed, filter the solution.
- Add Iodine.
- Add potassium iodide.
- Add diluted ammonia.
- If no blue colour is observed, heat carefully to boiling. In the presence of acetates, a dark color develops or a blue precipitate is formed.
Science behind.
Not much is known about the mechanism of the reaction or the nature of the blue complex formed (source 1). The reaction has been known since 1857 and its sensitivity and specificity studied (see for example reference 2).
Following the USP, it should be noted that in an alkaline medium, the lanthanum might form insoluble lanthanum hydroxide (La(OH)3), that can be separated by filtration.
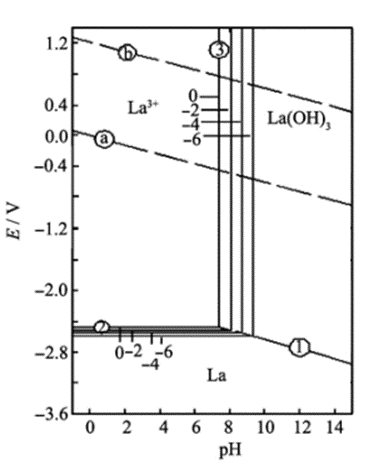
Pourbaix diagram for lanthanum-water. Source: Study on lanthanum salt conversion coating modified with citric acid on hot dip galvanized steel (sciencedirectassets.com)
The addition of potassium iodide (KI) in the solution helps to solubilize the iodine (I2), which has relatively poor solubility in water, by the formation of potassium triiodide (KI3). Unlike I2, I3− salts can be highly water-soluble (sources 1 and 5).
Sources:
- Ole Pederson. “Pharmaceutical Chemical Analysis”. (CRC press Taylor and Francis group. 2006).
- The lanthanum nitrate test for acetate in inorganic qualitative analysis (ias.ac.in)
- Lanthanum hydroxide – Wikipedia
- Study on lanthanum salt conversion coating modified with citric acid on hot dip galvanized steel (sciencedirectassets.com)
- Potassium iodide – Wikipedia
Reaction with iron (III) chloride.
This is reaction 3 as per JP <1.09> and reaction B as per USP <191>.
Procedure:
- The sample has to be prepared as a neutral solution.
- The addition of iron (III) chloride produces a red-brown solution. JP also indicates that a red-brown precipitate is formed upon boiling the red-brown solution.
- JP: The addition of hydrochloric acid dissolves the precipitate and changes the colour of the solution to yellow.
- USP: The red colour of the solution is destroyed by the addition of mineral acids.
Science behind.
The iron (III) chloride (FeCl3) reacts with the acetate (CH3-COO–) to form the red-brown complex of iron (III) acetate [Fe3O(CH3COO−)6(H2O)3]+:
3Fe+3 + 6CH3COO− + H2O ⇄ [Fe3(OH)2(CH3COO)6]+ + 2H+
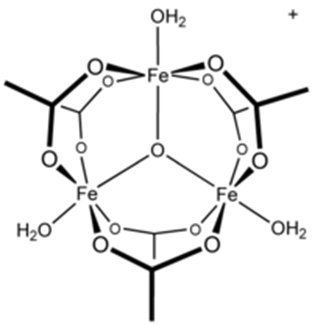
Structure of the complex iron (III) acetate. Source: Iron(III) acetate – Wikipedia
A neutral solution is required to allow the acetate to be as acetate form (CH3-COO–) and not as acetic acid (CH3-COOH) to speed the reaction.
The formation of the red-brown precipitate when boiling the solution is likely due to the hydrolysis of iron (III) chloride to form iron (III) hydroxide (source 3):
FeCl3 + 3H2O → Fe(OH)3↓ + 3HCl
The addition of an acid will destroy the colour of the solution by displacing to the left the equilibrium of the reaction:
3Fe+3 + 6CH3COO− + H2O ⇄ [Fe3(OH)2(CH3COO)6]+ + 2H+
In the case of the JP, where the acid is hydrochloric acid (HCl), the precipitate of Fe(OH)3 is dissolved by the HCl to form iron (III) chloride that after hydrolysis forms a yellow acidic solution (source 4):
Fe(OH)3 + 3HCl → FeCl3 + H2O
Sources:
- Iron(III) acetate – Wikipedia.
- solubility – Why is it advised to use neutral ferric chloride solution while performing confirmatory test for acetate ions? – Chemistry Stack Exchange.
- The formation of a sol | Experiment | RSC Education.
- The hydrolysis of iron(III) chloride | MEL Chemistry (melscience.com)
Acetone
In the European Pharmacopoeia (EP), acetone is identified using wet chemistry techniques through reactions b (reaction with nitroprusside) and c (reaction with nitrobenzaldehyde), identification a is refers to its relative density. On the other hand, the United States Pharmacopoeia (USP) employs spectroscopy and chromatography for acetone identification. The Japanese Pharmacopoeia (JP) does not have identification test for acetone.
Reaction with nitroprusside (EP reaction b)
Procedure:
- To the sample add dilute sodium hydroxide solution and sodium nitroprusside solution. An intense red colour is produced.
- Add acetic acid, the colour changes to violet.
Science behind:
Structure of the sodium nitroprusside:
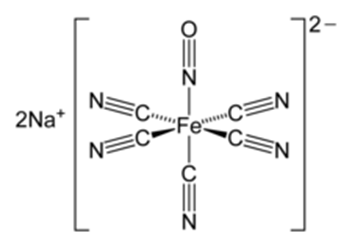
Source: Sodium nitroprusside – Wikipedia
This is a widely used identification test for ketones known as Legal test. The acetone (CH3-CO-CH3) is ionized by the sodium hydroxide (NaOH) and this ion reacts with the sodium nitroprusside (Na2Fe(CN)6NO) to form an intense red-coloured complex:
CH3COCH3 + OH–→ CH3COCH2– + H2O
[Fe(CN)5NO]2- + CH3COCH2– → [Fe(CN)5NO.CH3COCH2]3-
The colour change to violet produced by the addition of acetic acid might be due to structural changes in the complex as a consequence of the pH change. I have not found details of the structures of the complexes, however, colour changes of this complex due to the pH were already reported by Le Nobel in 1884 (indicated in source 3).
Sources:
- https://byjus.com/chemistry/tests-for-aldehydes-and-ketones/
- Sodium nitroprusside – Wikipedia
- A. C. H. Rothera. J Physiol. 1908 Dec 15; 37(5-6): 491–494.
Reaction with nitrobenzaldehyde (EP reaction c)
Procedure:
- Prepare a 0.1 per cent V/V solution of acetone in ethanol (50 per cent V/V)
- Add a solution of 2-Nitrobenzaldehyde in ethanol (50 per cent V/V) and a strong sodium hydroxide solution.
- Allow to stand for about 2 min.
- Acidify with acetic acid. A greenish-blue colour is produced.
Science behind:
The small concentration of the solution (0.1% V/V acetone) is to ensure the sensitivity of the test.
A mixture of ethanol and water is used because the 2-Nitrobenzaldehyde is slightly soluble in water but soluble in ethanol (source 1).
The reaction between the acetone (CH3-CO-CH3) and the 2-Nitrobenzaldehyde (C7H5NO3) in alkaline medium is an aldol condensation known as Baeyer–Drewsen indigo synthesis:
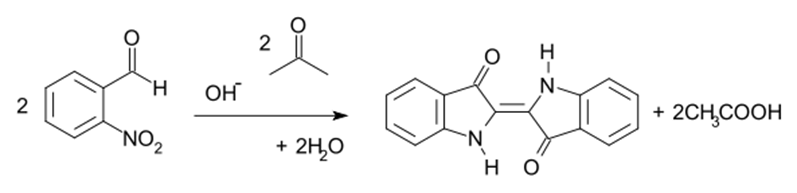
Source: Baeyer–Drewsen indigo synthesis – Wikipedia
The 2 minutes is required to allow the reaction to proceed.
The addition of acetic acid will lead to a decrease in the pH, leading to the reduction of indigo to Leuco-Indigo (source 3), giving a more greenish colour (source 4):
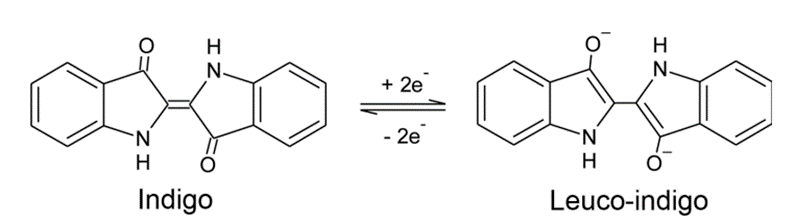
Sources:
- 2-Nitrobenzaldehyde | 552-89-6 (chemicalbook.com)
- Baeyer–Drewsen indigo synthesis – Wikipedia
- (PDF) Effect of pH Condition on Natural Indigo (Indigofera tinctoria) Reduction by Yeast (Saccharomyces cerevisiae) (researchgate.net)
- Frequently Asked Questions About Indigo – Botanical Colors
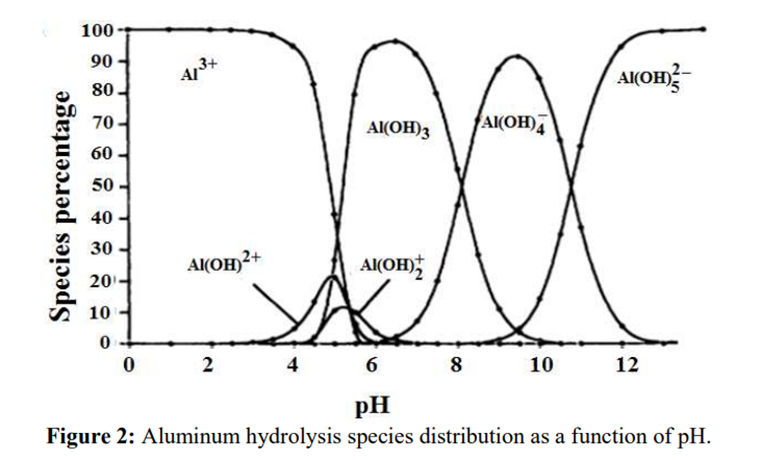
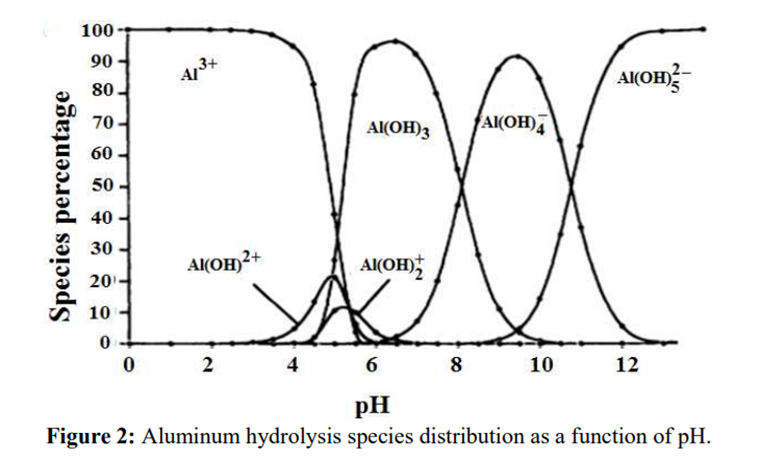
Aluminium
Aluminium is identified as the aluminium ion (Al³⁺) in EP (2.3.1), JP <1.09> and USP <191>. While some of the tests in these pharmacopoeias are quite similar and share the same scientific principles, there are some differences among them. Therefore, I address each test individually to account for these variations.
EP (2.3.1)
Procedure
- An aqueous solution of the sample is mixed with dilute hydrochloric acid and an aqueous solution of thioacetamide.
- Dropwise addition of dilute sodium hydroxide solution produces a gelatinous white precipitate.
- Further addition of dilute sodium hydroxide solution dissolves the precipitate.
- The gelatinous white precipitate is re-formed with the gradual addition of ammonium chloride solution.
Science behind
The identification reactions are based on the amphoteric behaviour of aluminium hydroxide (Al(OH)₃): this hydroxide is insoluble in alkaline conditions but dissolves in excess strong base such as sodium hydroxide.
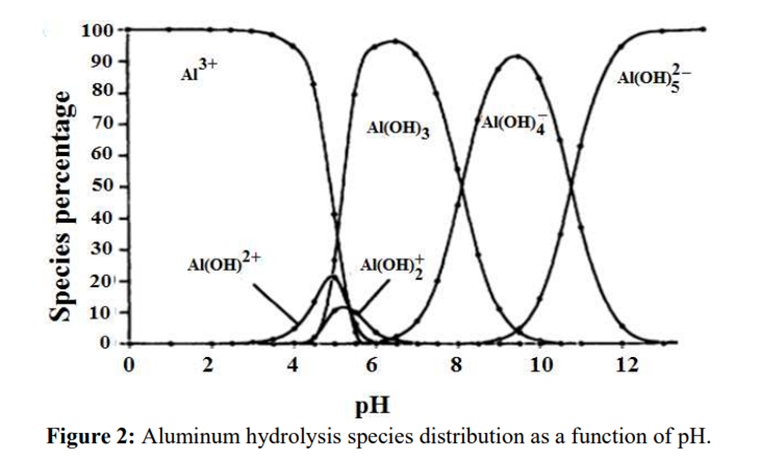
Source: B.Lekhlif, L. Oudrhiri, F. Zidane, P. Drogui, J. F. Blais, (2014), Journal of Materials and Environmental Sciences, 5 (1), pages 11-120. “Study of the electrocoagulation of electroplating industry wastewaters charged by nickel (II) and chromium (VI)” 13-JMES-510-2014-Lekhlif
The 1st step is used to eliminate by precipitation with sulfide (S²⁻) a significant number of cations (like Pb²⁺, Sn²⁺, Sn⁴⁺, and Sb³⁺) that also form amphoteric hydroxides. The use of thioacetamide (CH3-C(S)-NH2) in acid medium is used to produce sulfide ions (S2-) that will precipitate the sulfide of these ions, for example, in the case of lead:
Pb2++ S2– → PbS2↓
In presence of Al3+, the sulfide ions can precipitate aluminium sulfide (Al2S3), however, aluminium sulfide salt has a higher degree of solubility and it is instable in aqueous solution, hydrolysing to hydrated aluminium oxides/hydroxides, depending on the pH. There are open sources stating different reactions under different conditions:
Al2S3 + 6H+ → 2Al3+ + 3H2S
Al2S3 + 6 H2O → 2 Al(OH)3 + 3 H2S
In step 2, the pH of the solution is increased to produce the formation of a white gelatinous precipitate of aluminium hydroxide:
Al3+ + 3OH– → Al(OH)3↓
In step 3, the aluminium hydroxide is dissolved by the formation of a hydroxo complex:
Al(OH)3↓ + OH– → [Al(OH)4]–
In the last step, the precipitate of aluminium hydroxide is reformed by the addition of ammonium chloride solution, resulting in a reduction of the pH:
[Al(OH)4]– + NH4+ → Al(OH)3↓
Sources:
- Thioacetamide – Wikipedia
- Ole Pederson. “Pharmaceutical Chemical Analysis”. (CRC press Taylor and Francis group. 2006)
- Is Al2S3 Soluble or Insoluble? Exploring the Chemical Properties
- Aluminium sulfide – Wikipedia
- Aluminium sulfide – Sciencemadness Wiki
- B.Lekhlif, L. Oudrhiri, F. Zidane, P. Drogui, J. F. Blais, (2014), Journal of Materials and Environmental Sciences, 5 (1), pages 11-120. “Study of the electrocoagulation of electroplating industry wastewaters charged by nickel (II) and chromium (VI)” 13-JMES-510-2014-Lekhlif
JP <1.09>
The JP has 4 different tests.
Test 1:
Procedure
Solutions of aluminium salts, when treated with a solution of ammonium chloride and ammonia yield a gelatinous, white precipitate which does not dissolve in an excess of ammonia TS.
Science behind.
The pH of the solution is adjusted to produce a white gelatinous precipitate of aluminum hydroxide (Al(OH)₃). However, the ammonia solution does not achieve a sufficiently alkaline pH to dissolve the hydroxide.
Al3+ + 3OH– → Al(OH)3↓
Source: B.Lekhlif, L. Oudrhiri, F. Zidane, P. Drogui, J. F. Blais, (2014), Journal of Materials and Environmental Sciences, 5 (1), pages 11-120. “Study of the electrocoagulation of electroplating industry wastewaters charged by nickel (II) and chromium (VI)” 13-JMES-510-2014-Lekhlif
Test 2:
Procedure
Solutions of aluminium salts, when treated with a solution of sodium hydroxide, yield a gelatinous, white precipitate which dissolves in an excess of the reagent.
Science behind.
As in the case of step 2 and step 3 in the EP: the pH of the solution is adjusted to produce the formation of a white gelatinous precipitate of aluminium hydroxide:
Al3+ + 3OH– → Al(OH)3↓
A further increase in the pH will dissolve aluminium hydroxide by the formation of a hydroxo complex:
Al(OH)3↓ + OH– → [Al(OH)4]–
Source: B.Lekhlif, L. Oudrhiri, F. Zidane, P. Drogui, J. F. Blais, (2014), Journal of Materials and Environmental Sciences, 5 (1), pages 11-120. “Study of the electrocoagulation of electroplating industry wastewaters charged by nickel (II) and chromium (VI)” 13-JMES-510-2014-Lekhlif
Test 3:
Procedure
Solutions of aluminium salts, when treated with a solution of sodium sulfide, yield a gelatinous, white precipitate which dissolves in an excess of the reagent.
Science behind
I have not found a specific reaction for this test, my first thought was that aluminium sulfide (Al2S3):
Al3+ + S2- → Al2S3
However, the Al2S3 is sensitive to moisture, hydrolysing to hydrated aluminium oxides/hydroxides (Aluminium sulfide – Wikipedia). One of these possible hydroxide is aluminium hydroxide (Al(OH)3), a gelatinous, white precipitate:
Al2S3 + 6H2O = 2Al(OH)3 + 3H2S
Aqueous solutions of sodium sulfide, Na2S, are strongly alkaline (Sodium sulfide – Wikipedia), therefore an excess of sodium sulfide will increase the pH to dissolve the aluminium hydroxide (as in Test 2 above):
Al(OH)3↓ + OH– → [Al(OH)4]–
Source: B.Lekhlif, L. Oudrhiri, F. Zidane, P. Drogui, J. F. Blais, (2014), Journal of Materials and Environmental Sciences, 5 (1), pages 11-120. “Study of the electrocoagulation of electroplating industry wastewaters charged by nickel (II) and chromium (VI)” 13-JMES-510-2014-Lekhlif
Test 4:
Procedure
- Add ammonia to solutions of aluminium salts until a gelatinous, white precipitate is produced.
- Addition of an alizarin red solution changes the colour of the precipitate changes to red.
Science behind
Alizarin red forms complexes with aluminium (III) that are used for the quantitative determination of Al3+ (see for example (PDF) Quantitative determination of Al(III) ion by using Alizarin Red S including its microspheres optical sensing material)). As the complex is pH dependant, the addition of ammonia is used to control the pH:
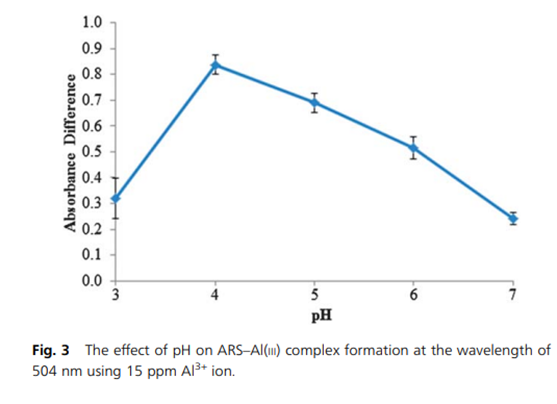
Source: Siti Mariam Supian, Tan Ling Ling, Lee Yook Heng and Kwok Feng Chong (2013) Analytical Methods 5, pages 2602 – 2609, “Quantitative determination of Al(III) ion by using Alizarin Red S including its microspheres optical sensing material”. (PDF) Quantitative determination of Al(III) ion by using Alizarin Red S including its microspheres optical sensing material.
Sources
- Aluminium sulfide – Wikipedia
- Sodium sulfide – Wikipedia
- B.Lekhlif, L. Oudrhiri, F. Zidane, P. Drogui, J. F. Blais, (2014), Journal of Materials and Environmental Sciences, 5 (1), pages 11-120. “Study of the electrocoagulation of electroplating industry wastewaters charged by nickel (II) and chromium (VI)” 13-JMES-510-2014-Lekhlif
- Siti Mariam Supian, Tan Ling Ling, Lee Yook Heng and Kwok Feng Chong (2013) Analytical Methods 5, pages 2602 – 2609, “Quantitative determination of Al(III) ion by using Alizarin Red S including its microspheres optical sensing material”. (PDF) Quantitative determination of Al(III) ion by using Alizarin Red S including its microspheres optical sensing material
USP <191>
The USP has two different tests.
Test A:
Procedure
Addition of ammonium hydroxide to solutions of solutions of aluminium salts yield a gelatinous, white precipitate that is insoluble in an excess of ammonium hydroxide.
Science behind.
The only ionic form of aluminium is aluminium (III). The addition of ammonia produces the precipitation of aluminium hydroxide, Al(OH)3:
Al3+ + 3OH– → Al(OH)3↓
But the pH is not alkaline enough to dissolve this amphoteric hydroxide:
Source: B.Lekhlif, L. Oudrhiri, F. Zidane, P. Drogui, J. F. Blais, (2014), Journal of Materials and Environmental Sciences, 5 (1), pages 11-120. “Study of the electrocoagulation of electroplating industry wastewaters charged by nickel (II) and chromium (VI)” 13-JMES-510-2014-Lekhlif
Test B
Procedure.
Addition of a solution of sodium hydroxide or sodium sulfide to solutions of aluminium salts produces a gelatinous, white precipitate, which dissolves in an excess of either of the same reagents.
Science behind.
In the case of the sodium hydroxide: the pH of the solution is adjusted to produce the formation of a white gelatinous precipitate of aluminium hydroxide:
Al3+ + 3OH– → Al(OH)3↓
A further increase in the pH will dissolve aluminium hydroxide by the formation of a hydroxo complex:
Al(OH)3↓ + OH– → [Al(OH)4]–
Source: B.Lekhlif, L. Oudrhiri, F. Zidane, P. Drogui, J. F. Blais, (2014), Journal of Materials and Environmental Sciences, 5 (1), pages 11-120. “Study of the electrocoagulation of electroplating industry wastewaters charged by nickel (II) and chromium (VI)” 13-JMES-510-2014-Lekhlif
In the case of sodium sulfide: I have not found a specific reaction for this test, my first thought was that aluminium sulfide (Al2S3):
Al3+ + S2- → Al2S3
However, the Al2S3 is sensitive to moisture, hydrolysing to hydrated aluminium oxides/hydroxides (Aluminium sulfide – Wikipedia). One of these possible hydroxide is aluminium hydroxide (Al(OH)3), a gelatinous, white precipitate:
Al2S3 + 6H2O ⇌ 2Al(OH)3 + 3H2S
Aqueous solutions of sodium sulfide, Na2S, are strongly alkaline (Sodium sulfide – Wikipedia), therefore an excess of sodium sulfide will increase the pH to dissolve the aluminium hydroxide (as in Test A above):
Al(OH)3↓ + OH– → [Al(OH)4]–
Source: B.Lekhlif, L. Oudrhiri, F. Zidane, P. Drogui, J. F. Blais, (2014), Journal of Materials and Environmental Sciences, 5 (1), pages 11-120. “Study of the electrocoagulation of electroplating industry wastewaters charged by nickel (II) and chromium (VI)” 13-JMES-510-2014-Lekhlif
Sources:
- Thioacetamide – Wikipedia
- Ole Pederson. “Pharmaceutical Chemical Analysis”. (CRC press Taylor and Francis group. 2006)
- B.Lekhlif, L. Oudrhiri, F. Zidane, P. Drogui, J. F. Blais, (2014), Journal of Materials and Environmental Sciences, 5 (1), pages 11-120. “Study of the electrocoagulation of electroplating industry wastewaters charged by nickel (II) and chromium (VI)” 13-JMES-510-2014-Lekhlif
- Aluminium sulfide – Wikipedia
- Sodium sulfide – Wikipedia
Ammonium
EP (2.3.1) differentiate between ‘ammonium salts’ and ‘ammonium salts and salts of volatile bases’, the JP<1.09> refers to ‘Ammonium salts’ and USP <191> refers to ‘Ammonium’. The ID tests in these pharmacopoeias can be classified as:
- Reaction with magnesium oxide (EP(2.3.1): ‘ammonium salts’ and USP<191>).
- Reaction with sodium hydroxide (EP(2.3.1): ‘ammonium salts and salts of volatile bases’ and JP<1.09>).
Reaction with magnesium oxide
EP (2.3.1): ‘ammonium salts’ and USP <191>.
Procedure
None of the pharmacopeia specify the glassware for this test, like it is required for the limit test of arsenic, but it will be required a couple of test tubes like this assembly:
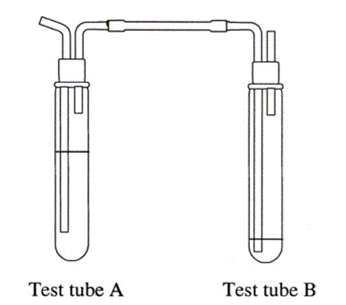
Source: Ole Pederson. “Pharmaceutical Chemical Analysis”. (CRC press Taylor and Francis group. 2006)
- The sample is prepared as an aqueous solution.
- Add magnesium oxide.
- Pass a current of air through the mixture and direct the gas that escapes just beneath the surface of a mixture of diluted hydrochloric acid and a methyl red solution. The indicator changes to yellow.
- The addition of a solution of sodium cobaltinitrite to the yellow solution produces a yellow precipitate.
Science behind
Magnesium oxide (MgO) is a moderately strong base with low solubility in water. When added to water, it reacts to form magnesium hydroxide (Mg(OH)₂), which only partially dissolves, making the solution weakly alkaline with a pH of around 9 to 10, depending on the conditions:
MgO + H2O → Mg(OH)2
The ammonium ions (NH4+) of the sample reacts with the hydroxide ion (OH–) of the magnesium hydroxide to produce ammonia (NH3) gas:
NH4+ + OH– → NH3↑ + H2O
As the magnesium hydroxide is a weaker base than the sodium hydroxide, it only liberates ammonia from ammonium salts and is not capable of hydrolysing amides and other nitrogen-bearing organic molecules as is the case of the reaction with ‘Reaction with sodium hydroxide’ (source 1).
The sodium cobaltinitrite (Na₃[Co(NO₂)₆]) added react with the ammonia (NH4+) forming yellow ammonium cobaltinitrite ((NH4)3[Co(NO2)6]):
3 NH4+ + Na₃[Co(NO₂)₆] → (NH4)3[Co(NO2)6]↓ + 3Na+
Sources
- Ole Pederson. “Pharmaceutical Chemical Analysis”. (CRC press Taylor and Francis group. 2006).
- Magnesium oxide – Wikipedia
- Sodium hexanitritocobaltate(III) – Wikipedia
Reaction with sodium hydroxide
EP (2.3.1): ‘ammonium salts and salts of volatile bases’ and JP<1.09>.
Procedure
- Prepare the sample as an aqueous solution.
- Add diluted sodium hydroxide.
- Heat the solution.
- The vapours produced alkaline reaction in pH paper.
Science behind
As in the case of the ‘Reaction with magnesium oxide’, the hydroxide ion (OH–) from the sodium hydroxide (NaOH) reacts with the ammonium ion (NH4+) to produce ammonia (NH3):
NH4+ + OH– → NH3↑ + H2O
But as the sodium hydroxide is a stronger base than the magnesium hydroxide (produced by the magnesium oxide in water), when heated, volatile nitrogen bases will also give an alkaline reaction due to the production of ammonia (like in the case of the ethanamide (CH3CONH2)) or amines (like the reaction with N-methylethanamide (CH3CONHCH3) that will produce methylamine (CH3NH2)):
CH3CONH2 + NaOH → CH3COONa + NH3↑
CH3CONHCH3 + NaOH → CH3COONa + CH3NH2↑
Sources
- Ole Pederson. “Pharmaceutical Chemical Analysis”. (CRC press Taylor and Francis group. 2006).
- Notes – 20 Nitrogen Compounds – CIE Chemistry A Level (physicsandmathstutor.com)
Bromide
There are 3 ID tests of bromide in EP (2.3.1), JP <1.09> and USP <191>, these are:
- Precipitation with silver (present in the 3 pharmacopeias).
- Reaction with fuchsin (EP).
- Reaction with chlorine solution (JP and USP).
Precipitation with silver.
Procedure
There are variations between the different pharmacopoeias, but we can summarize the steps as:
- Prepare an aqueous solution with the sample.
- EP prescribes acidification with dilute nitric acid, USP and JP does not state acidification at this stage, but it is indicated after the next step.
- Addition of silver nitrate solution a yellowish precipitate.
- The precipitate is partially soluble in ammonia.
- The precipitate is insoluble in dilute nitric acid.
Science behind.
The science behind this ID test is similar to the ones of chloride and iodide. These three anions are distinguished by the colour of the silver precipitate formed and their solubility in ammonia:
| Precipitate | Colour | Solubility in ammonia |
|---|---|---|
| AgCl | White | Soluble in excess of ammonia |
| AgBr | Pale yellow, creamy | Partly soluble |
| AgI | Yellow | Insoluble |
The addition of silver nitrate (AgNO3) leads to the precipitation of pale-yellow silver bromide:
Ag+ + Br– → AgBr↓
The washing of the precipitate requires subdued light when the precipitate is exposed to light turns grayish or black as Ag+ is reduced to metallic silver.
2ABr↓ + λ → 2Ag↓ + Cl2↑
The precipitated AgBr is partially soluble in excess of ammonia due to the complexation of the silver by ammonia to form diamminoargentate complex:
AgBr↓ + 2NH3 ⇄ [Ag(NHl3)2]+ + Br–
The solubility of silver bromides is intermediate compared to that of silver chloride and silver iodide. So the complex formation constant of the diammninoargentate complex overrules the solubility constant of silver chloride easily, and with difficulty the solubility constant of silver bromide, but it is not able to do so in the case of silver iodide (source 2).
The presence of nitric acid excludes a large number of substances. One group is the silver salts of weak acids that are unstable in dilute nitric acid and decompose to gas, such as carbonate and sulfite.
Sources
- Silver bromide – Wikipedia
- Ole Pederson. “Pharmaceutical Chemical Analysis”. (CRC press Taylor and Francis group. 2006)
- inorganic chemistry – What is the reason for the different solubility of silver halides in ammonia? – Chemistry Stack Exchange
Reaction with fuchsin.
Fuchsin is also known as rosaniline hydrochloride or Schiff’s reagent. This reaction is only used in EP.
Procedure
- To an aqueous solution of the sample, add lead dioxide, acetic acid and shake gently.
- Dry the inside of the upper part of the test-tube with a piece of filter paper and allow to stand for 5 min.
- Impregnate a piece of filter paper by dipping the tip in a drop of decolorised fuchsin solution and introduce the impregnated part immediately into the tube.
- Starting from the tip, a violet colour appears within 10 s that is clearly distinguishable from the red colour of fuchsin, which may be visible on a small area at the top of the impregnated part of the paper strip.
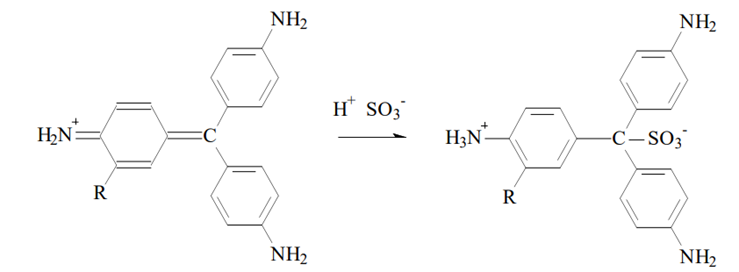
Science behind
The addition of lead oxide (PbO2) and acetic acid oxidize the bromide (Br–) to bromine (Br2):
2Br– + PbO2 + 4H+ → Br2 + Pb2+ + 2H2O
The decolorized fuchsin solution is a mixture of rosaniline hydrochloride and para-rosaniline hydrochloride decolorised by the addition of sodium sulfite (Na2SO3).
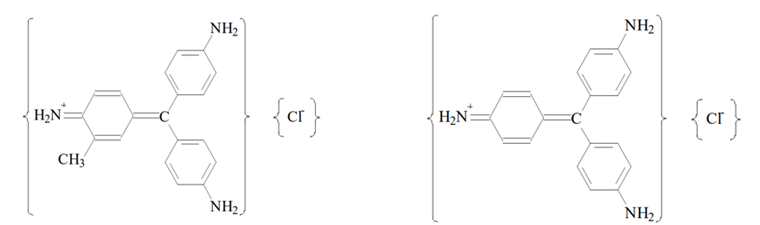
Rosaline hydrochloride (left) and para rosaline hydrochloride. Source: Ole Pederson. “Pharmaceutical Chemical Analysis”. (CRC press Taylor and Francis group. 2006).
Decolouration. Source: Ole Pederson. “Pharmaceutical Chemical Analysis”. (CRC press Taylor and Francis group. 2006).
The decolorised fuchsin reacts with the bromine (Br2) to form violet pentabromine rosaline and hexabromine rosaline:
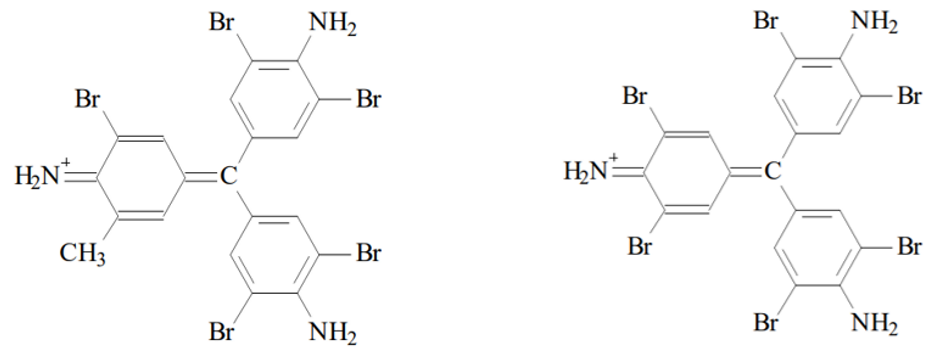
Pentabromine rosaline (left) and Hexabromine rosaline (right). Source: Ole Pederson. “Pharmaceutical Chemical Analysis”. (CRC press Taylor and Francis group. 2006).
The red colour that might be appear in the paper is due to the formation of the original solution of fuchsin produced by heat.
This is selective for bromine as neither chlorine nor iodide colorizes decolorized fuchsin. Additionally, chlorine is not oxidized by lead oxide.
Sources
- Ole Pederson. “Pharmaceutical Chemical Analysis”. (CRC press Taylor and Francis group. 2006)
- Schiff test – Wikipedia
Reaction with chlorine solution
This reaction appears in the USP and in the JP.
Procedure
- Solutions of bromides are mixed with a saturated solution of chlorine produces bromine (identified by a yellow-brown colour).
- A portion of the solution obtained in the 1st step is mixed with chloroform. A red to brown colour is formed.
- To another portion of the solution obtained in the 1st step is added phenol, a white precipitate is formed (this step appears only in the JP)
Science behind
The addition of chlorine (Cl2) to a solution with bromides (Br–) produces bromine (Br2) as a displacement reaction:
Cl2 + 2Br– → Br2 + 2Cl–
The bromide is soluble in chloroform, where it forms a reddish-brownish solution.
The addition of phenol produces 2,4,6-tribromophenol:
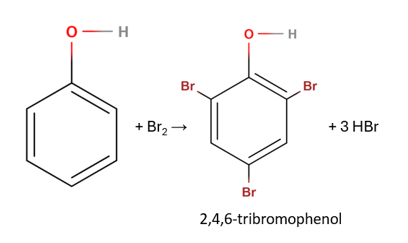
Sources
- Group 7 (VII) displacement reactions – Higher tier – The periodic table – (CCEA) – GCSE Combined Science Revision – CCEA Double Award – BBC Bitesize
- Bromine | Br2 | CID 24408 – PubChem (nih.gov)
- Reaction of Chlorine with Potassium Bromide (chemedx.org)
- Ring Reactions of Phenol – Chemistry LibreTexts
Chloride
There are 3 ID tests of chloride mentioned in EP (2.3.1), USP <191> and JP <1.09>, these tests are:
- Precipitation with silver (present in the 3 pharmacopeia)
- Reaction with potassium dichromate and sulfuric acid (only EP)
- Reaction with potassium permanganate and sulfuric acid (only JP)
Precipitation with silver
Procedure as per EP and USP:
- The sample is dissolved in water
- Solution is acidified with nitric acid and silver nitrate is added. A curdled white precipitate of silver chloride is formed.
- Precipitate is centrifugate and washed with water in subdue light.
- The precipitate is suspended in water and dissolved with ammonia.
The JP procedure is slightly different approach, but the principles are the same:
- Sample is dissolved in water
- Addition of silver nitrate yields a white precipitate.
- A portion of the precipitate is treated with dilute nitric acid and the precipitate does not dissolve.
- Another portion of the precipitate is treated with excess of ammonia and the precipitate dissolves.
The science behind
The science behind this ID test is similar to the ones of bromide and iodide. These three anions are distinguished by the colour of the silver precipitate formed and their solubility in ammonia:
The addition of silver nitrate (AgNO3) leads to the precipitate of white silver chloride:
| Precipitate | Colour | Solubility in ammonia |
|---|---|---|
| AgCl | White | Soluble in excess of ammonia |
| AgBr | Pale yellow, creamy | Partly soluble |
| AgI | Yellow | Insoluble |
Ag+ + Cl– → AgCl↓
The presence of nitric acid excludes a large number of substances. One group is the silver salts of weak acids that are unstable in dilute nitric acid and decompose to gas, such as carbonate and sulfite.
The precipitated AgCl is dissolved in excess of ammonia due to the complexation of the silver by ammonia to form diamminoargentate complex:
AgCl↓ + 2NH3 ⇄ [Ag(NH3)2]+ + Cl–
It should be noted that AgCl is very stable, therefore it is required to have an excess of ammonia to displace the equilibrium to the right (Le-Chatelier principle). AgBr and AgI will not dissolve in excess of ammonia as their solubility products are smaller than that of the AgCl.
One last note about this reaction: the washing of the precipitate requires subdued light when the precipitate is exposed to light turns grayish or black as Ag+ is reduced to metallic silver.
2AgCl↓ + λ → 2Ag↓ + Cl2↑
Reaction with potassium dichromate and sulfuric acid
This is reaction is only performed in the EP.
Procedure
- In a test tube the sample is mixed with potassium dichromate and sulfuric acid.
- Filter-paper strip impregnated with a solution of diphenylcarbazide is placed over the opening of the test-tube turns violet-red. The impregnated paper must not come into contact with the potassium dichromate.
Science behind
In the presence of chloride and sulfuric acid, the dichromate (Cr2O72-) is converted into chromyl chloride CrO2Cl2 gas:
4Cl– + Cr2O72– + 6H+ → 2CrO2Cl2↑ + 3H2O
The chromium (VI) of chromyl chloride oxidizes diphenylcarbazide to diphenylcarbazone and chromium (III) is produced. The chromium (III) and the formed diphenylcarbazone form a strong-coloured complex.
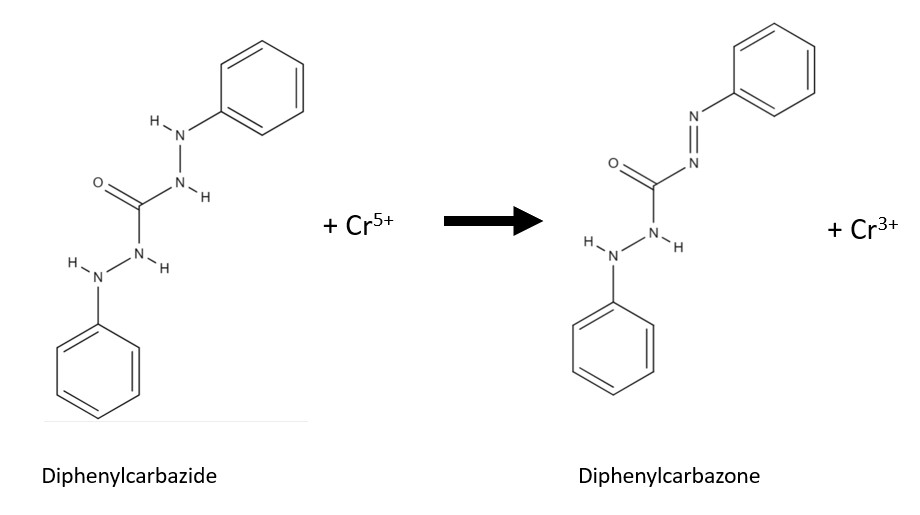
The impregnated paper must not come into contact with the potassium dichromate because the potassium dichromate is a chromium(VI) compound like chromyl chloride and will produce similar rection.
With the exception of fluoride, this test is said to have a high degree of selectivity.
Reaction with sulfuric acid and potassium permanganate
This test is only performed in the JP.
Procedure
- In a test tube the sample is mixed with potassium permanganate and sulfuric acid.
- Filter-paper strip impregnated with a solution of potassium iodide starch placed over the opening of the test-tube turns blue.
Science behind
In an acid medium, the permanganate (MnO42‑) oxidises the chloride (Cl‑) to chlorine (Cl2) gas:
8H+ + MnO42- + 4Cl– → 2Cl2↑ + Mn2+ + 4H2O
The chlorine (Cl2) reacts with the potassium iodide (KI) to form iodine (I2) that will turn blue the starch:
Cl2 + 2KI → I2 + 2KCl
Sources:
- Ole Pederson. “Pharmaceutical Chemical Analysis”. (CRC press Taylor and Francis group. 2006)
- inorganic chemistry – What is the reason for the different solubility of silver halides in ammonia? – Chemistry Stack Exchange
Iodide
There are 4 different ID tests for iodides in the EP (2.3.1), JP<1.09> and USP<191>:
- Precipitation with silver nitrate (EP, JP and USP).
- Reaction with potassium dichromate (EP only).
- Precipitation with of sodium nitrite (JP only).
- Reaction with chlorine (USP only)
Precipitation with Silver Nitrate.
Procedure
- Aqueous solution is prepared with the sample. EP requires acidification with dilute nitric acid.
- Silver nitrate solution is added. A yellow precipitate is formed.
- This precipitate is not dissolved in ammonia and it also not dissolved in nitric acid.
Science behind
The test is identical to identification test used for chlorides and bromides. These three anions are distinguished by the colour of the silver precipitate formed and their solubility in ammonia:
| Precipitate | Colour | Solubility in ammonia |
|---|---|---|
| AgCl | White | Soluble in excess of ammonia |
| AgBr | Pale yellow, creamy | Partly soluble |
| AgI | Yellow | Insoluble |
The addition of silver nitrate (AgNO3) leads to the precipitate of yellow silver iodide:
Ag+ + I– → AgI↓
The presence of nitric acid excludes a large number of substances. One group is the silver salts of weak acids that are unstable in dilute nitric acid and decompose to gas, such as carbonate and sulfite.
Contrary to chloride and partly bromide, the silver iodide does not dissolve in ammonia because its low solubility product prevents its dissolution with ammonia, despite the formation of complexes of ammonia with silver (see chloride limit test).
One last note about this reaction: the washing of the precipitate requires subdued light when the precipitate is exposed to light turns grayish or black as Ag+ is reduced to metallic silver:
2AgI↓ + λ → 2Ag↓ + Br2↑
Sources
- Ole Pederson. “Pharmaceutical Chemical Analysis”. (CRC press Taylor and Francis group. 2006)
- inorganic chemistry – What is the reason for the different solubility of silver halides in ammonia? – Chemistry Stack Exchange
- Silver halide – Wikipedia
Reaction with potassium dichromate
Procedure
- Prepare and aqueous solution with the sample.
- Add dilute sulfuric acid, potassium dichromate in solution water and chloroform.
- Shake and allow to stand.
- The chloroform layer is coloured violet or violet-red.
Science behind
The iodide (I–) in acid medium is oxidized by the dichromate to iodine (I2).
6I– + Cr2O72– +7H2SO4 → 3I2 + 2Cr3+ + 7SO42– +7H2O
The iodine produced is more soluble in the (lower) chloroform phase, in which it is easily recognized by the colour.
Sources
- Ole Pederson. “Pharmaceutical Chemical Analysis”. (CRC press Taylor and Francis group. 2006)
Precipitation with of sodium nitrite
Procedure
- Prepare an acidic aqueous solution with the sample.
- Addition of sodium nitrite solution produces a yellow-brown colour followed by a black-purple precipitate.
- Addition of starch solution produces a deep blue colour.
Science behind
A video with this reaction can be seen in source 1.
The sodium nitrite (NaNO2) in acidic solution oxide the iodide (I–) to iodine (I2):
2I‑ + 2NO2– + 4H+ → I2 + 2NO + 2H2O
The initial colour (yellow-brown) is due to the formation of triiodide (I3–) from the I2 and the I–:
I2 + I– → I3–
As the colour triiodide solution varies from yellow (low concentration of I3–) to brown (high concentration of I3–). Eventually, I2 is produced and precipitate (black-purple precipitate).
The addition of starch solution is due to the formation of chains of amylose where the iodine is fitted.
Sources
- Sodium Nitrite and Potassium Iodide ( Reaction ) (youtube.com)
- Triiodide – Wikipedia
- Why Does Iodine Turn Starch Blue? – ChemistryViews
Reaction with chlorine
Procedure
- Aqueous solutions of iodides, upon dropwise addition of a saturated solution of chlorine, liberate iodine, which colours the solution from yellow to red.
- When the solution is shaken with chloroform, the solution is coloured violet.
- The iodine liberated gives a blue colour with starch TS.
Science behind
The addition of chlorine (Cl2) to a solution with iodide (Br–) produces iodine (I2) as a displacement reaction:
Cl2 + 2I– → I2 + 2Cl–
Small additions of chlorine will produce the gradual formation of iodine that will range from yellow to red due to the formation of triiodide (I3–):
I2 + I– → I3–
An excess of chlorine solution will produce a dark precipitate of bromide.
Iodine dissolves easily in chloroform to form a solution.
The addition of starch solution is due to the formation of chains of amylose where the iodine is fitted.
Sources
- Halogens as oxidising agents (chemguide.co.uk)
- Triiodide – Wikipedia
- Iodine Testing | Elemental Testing and Analysis | Midwest Microlab (midwestlab.com)
- Why Does Iodine Turn Starch Blue? – ChemistryViews
Phosphate (orthophosphate)
The clarification of orthophosphates indicate that the reaction is applicable to single PO42- units. The term phosphate includes, not only the orthophosphates (PO42-), but also any structure composed of PO42- units, such as pyrophosphates (PO3-O-PO34-), triphosphorates (PO3-O-PO2-O-PO35-), etc.
The two first limit tests of orthophosphates as per EP (2.3.1), JP <1.09> and USP <191> are very similar in the 3 pharmacopeias. JP has an additional test. These tests are:
- Precipitation with silver nitrate (1st reaction in EP, JP and USP)
- Precipitation with ammonium molybdate (2nd reaction in EP, JP and USP)
- Precipitation with magnesium ions (3rd reaction in JP)
Precipitation with silver nitrate
Procedure:
- To a neutral solution of phosphates, addition of a silver nitrate solution produces a yellow precipitate. EP also indicates that the colour is not changed by boiling (no mentioned in USP or JP).
- The yellow precipitate is dissolved by the addition of ammonia.
- The yellow precipitate formed in the first step is dissolved by the addition of dilute nitric acid (this step appears only in JP and USP)
Science behind:
This reaction has limited selectivity and it might require additional testing. Neutral solution is required to eliminate possible secondary reactions that will prevent the formation of the yellow precipitate, like the reaction with ammonia or with nitric acid indicated below.
The addition of silver nitrate (AgNO3) to the phosphate (PO43-) solution produces a yellow precipitate of silver nitrate (AgNO3):
3Ag+ + HPO42- → Ag3PO4↓ + H+
As per EP indicates, boiling does not change colour. This is used to distinguish from phosphorous acid (H3PO3) and hypophosphorous acid (H3PO2) which would reduce the silver ion (Ag+) to metallic silver, turning the precipitate black. I have only found details of the reaction silver salt of the phosphorous acid (silver phosphite (Ag2HPO3)):
Ag2HPO3 + H2O → H3PO4 + 2Ag↓
Addition of ammonia (NH3) dissolves the precipitate due to the formation of silver ions with ammonia:
Ag3PO4↓ + 6NH3 → 3[Ag(NH3)2]+ + PO43-
The addition of dilute nitric acid (HNO3) dissolves the precipitate to produce silver nitrate (in solution) and phosphoric acid.
3HNO3 + Ag3PO4↓ → H3PO4 + 3Ag+ + 3NO3–
Sources:
- https://www.toppr.com/ask/question/silver-phosphite-ag2hpo3-is-warmed-with-water-and-the-silvery-substance-is-precipitated/
- Silver Phosphate Formula, Structure, Properties, Uses (pw.live)
- Ole Pederson. “Pharmaceutical Chemical Analysis”. (CRC press Taylor and Francis group. 2006)
Precipitation with ammonium molybdate
Procedure:
- Acidify the sample solution (JP mention acidify with nitric acid, USP only say acid solutions and EP does not mention acidification referring to the ‘prescribed solution’).
- Add ammonium molybdite reagent. A yellow precipitate forms. USP states that ‘The precipitate may be slow to form’ and the JP requires warming.
- The yellow precipitate is dissolved by the addition of ammonia (JP and USP) or sodium hydroxide (JP).
Science behind:
I have not found a clear answer to the science behind this reaction. As the source 1 mention, “the nature of the substance formed has not been fully elucidated and it has been referred as an acid, a complex and in some cases a salt”. As example, this source mentions:
(NH4)3PO4 · NH4VO3 ·16MoO3
Sources 2 and 3 mention that the yellow precipitate is due to the formation of ammonium phosphomolybdate ((NH4)3PO4 ⋅ 12MoO3):
H3PO4 + 12(NH4)2MoO4 + 21HNO3 → (NH4)3PO4 ⋅ 12MoO3 + 21NH4NO3 + 12H2O
Sources 4 indicates that the yellow precipitate is due to the formation vanadomolybdophosphoric acid, but I have not found a reliable resource with its formula (despite it is mentioned in several publications).
Sources:
- Ole Pederson. “Pharmaceutical Chemical Analysis”. (CRC press Taylor and Francis group. 2006)
- Phosphate radical with ammonium molybdate gives precipitate of which colour (toppr.com)
- Phosphate test – Wikipedia
- Determination of Phosphate by a Colorimetric Method – Chemistry LibreTexts
Precipitation with magnesium ions
Only mentioned in JP
Procedure:
- To a neutral or ammonia-alkaline solution of phosphates add magnesium (II) solution. A white, crystalline precipitate is formed.
- Addition of dilute hydrochloric dissolves the precipitate
Science behind
Magnesium ions (Mg2+) are not precipitated by phosphate ions (PO43-) under acid conditions. Under neutral conditions, it is precipitated as magnesium hydrogen phosphate (MgHPO4)
Mg2+ + HPO42− → MgHPO4↓
In the presence of ammonia, magnesium ammonium phosphate (MgNH4PO4) is precipitated:
Mg2+ + NH3 + HPO42− → MgNH4PO4↓
The addition of hydrochloric acid (HCl) produces a double displacement reaction: the magnesium ion (Mg²⁺) from the precipitate reacts with the chloride ion (Cl⁻) from HCl to form magnesium chloride (MgCl2), that dissolves, and the hydrogen ion (H⁺) from HCl reacts with the phosphate ion (either HPO4²⁻ or NH4PO42-) of the precipitate to form phosphoric acid (H3PO4):
MgHPO4↓ + 2HCl → Mg2+ + 2Cl– + H3PO4
Or:
MgNH4PO4↓ + 3HCl → Mg2+ + 3Cl– + NH4+ + H3PO4
Sources
Propylene glycol (JP, ID 1)
Thank you to Harry Lawrence (Harry.Lawrence@rssl.com) for helping with this ID test.
Procedure
- Mix propylene glycol with triphenylchloromethane (also known as trityl chloride), pyridine and heat under reflux for 1 hour.
- After cooling, dissolve the mixture in acetone by warming, shake with activated charcoal and filter.
- Concentrate the filtrate and cool. Collect the separated crystals and dry in a desiccator. The crystals melt between 174°C and 178°C.
Science behind
The only information that I have found about this reaction (see source 1) is that following this procedure, “trityl ether of propylene glycol” is obtained, with formula C41H36O2 and a melting point of 176.5 – 177.0°C.
What follows is my understanding of what might be happening based on general chemistry and the information from source 1.
Using propylene glycol (CH3-CHOH-CH2OH) as a starting point and with a final formula C41H36O2, we have two possibilities 1,3-Bis(triphenylmethoxy)propane and 1,2-Bis(triphenylmethoxy)propane (or maybe a mixture of both). I’m more inclined to 1,3-Bis(triphenylmethoxy)propane due to steric hindrance from the phenyl groups (-C5H6), however, I have no data to back this statement.
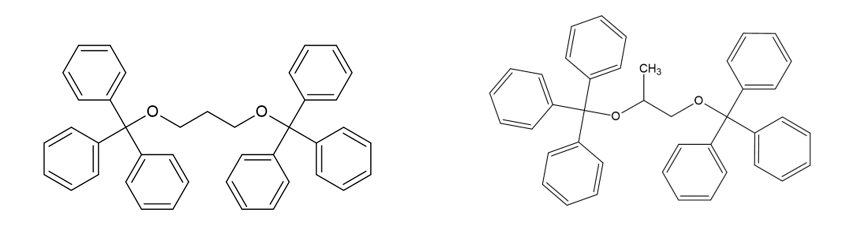
1,3-Bis(triphenylmethoxy)propane (left) and 1,2-Bis(triphenylmethoxy)propane (right)
The reaction is a Williamson ether synthesis where the pyridine (C5H5N) acts as a base allowing the etherification of the propylene glycol (CH3-CHOH-CH2OH) by the trityl chloride Cl-C(C6H6)3. The possible reactions are:
1. For 1,2-Bis(triphenylmethoxy)propane, the reaction seems more straightforward on paper:
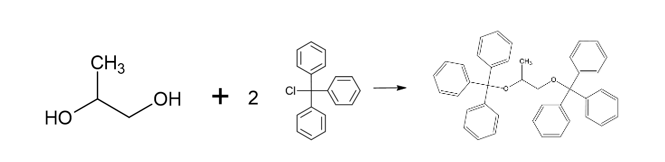
2. For 1,3-Bis(triphenylmethoxy)propane, my best guess is that it should be an intermediate that allows to transfer the of oxygen from the carbon 2 to carbon 3, something like:
And the reaction would be something like:
The implementation of a one-hour reflux procedure is intended to facilitate the progression of the reaction. This is particularly crucial in this context due to the presence of voluminous phenyl groups, which could potentially induce steric hindrance, thereby inhibiting the reaction from proceeding efficiently.
Charcoal and acetone are used to remove impurities and concentrate the final product.
Sources
- Nancy Green and Melvin W. Green, “The Preparation of the Trityl Ether of Propylene Glycol” Journal of the American Chemical Society. (1944), 66, 9, 1610–1611
- Williamson Ether Synthesis – Chemistry LibreTexts
- Williamson ether synthesis – Wikipedia
- Pyridinium – Wikipedia
- Pyridine – Wikipedia
Sodium
There are 3 ID tests mentioned in EP (2.3.1), USP <191> and JP <1.09>, these tests are:
- Reaction with pyrantimonate (present in the 3 pharmacopeia)
- Reaction with methoxyphenylacetic (only EP)
- Flame test (only JP)
Reaction with Pyroantimonate
Procedure as per EP and USP:
- The sample is dissolved in water
- Potassium carbonate is added and heated to boiling. No precipitate is formed.
- Potassium pyroantimonate is added, heated to boiling, cooled in iced water and a dense precipitate is formed (the formation of the precipitate might require to rub the inside of the test tube with a glass rod.
JP follow similar procedure, but it requires that the sample has been previously concentrated and made neutral or slightly alkaline, however, the general chapter does not specify how to reach these conditions. The wording of the JP is: “Neutral solutions of sodium salts yield a white, crystalline precipitate with potassium hexahydroxoantimonate (V) TS. The formation of the precipitate is accelerated by rubbing the inside wall of the test tube with a glass rod.”
Science behind:
There are two reasons for adding potassium carbonate:
- Increases the pH of the solution, preventing antimonate precipitates as antimonic acid (H3SbO4), a reaction that could occur in neutral and weakly acidic solutions.
Sb5+ + 4H2O → H3SbO4↓ + 5H+
2. Reveal the presence of cations, like calcium, that would otherwise give a false positive reaction by precipitating as carbonates.
In the presence of sodium, the addition of pyroantimonate solution (KSb(OH)6) form sodium antimonate:
Na+ + [Sb(OH)6]– → Na[Sb(OH)6]↓
This salt has a tendency of forming rather stable super-saturated solutions, and this is the reason the test solution has to be cooled down and precipitation initiated by rubbing with a glass rod.
Reaction with methoxyphenylacetic
Only in EP
Procedure
- The sample is dissolved in water.
- Methoxyphenylacetic reagent is added and cooled in ice-water for 30 min. A voluminous, white, crystalline precipitate is formed.
- Placed in water at 20°C and stir for 5 min. The precipitate does not disappear.
- The precipitate dissolved completely by the addition of diluted ammonia.
- Addition of ammonium carbonate do not form a precipitate.
The science behind.
The precipitate is the sodium salt of methoxyphenylacetic acid:
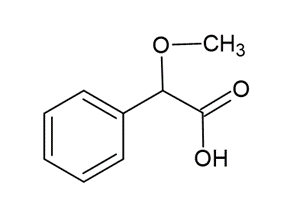
Methoxyphenylacetic acid
The precipitation has an optimum pH interval 3 to 4 (achieved by the reagent).
Heating at 20°C will dissolve other alkalis that also precipitate the relevant salt with methoxyphenylacetic acid.
The increase of pH due to the addition of ammonia and the ammonium carbonate dissolves the sodium salt, but will not dissolve the salts of calcium, strontium, or barium, since these ions form insoluble hydroxides and carbonates at high pH.
Flame test
The JP chapter <1.09> indicates that the Flame Coloration Test (1) <1.04> gives a yellow colour. The science behind the flame test is that when the compound is heated, the metal ions gain energy and move from a lower to a higher energy level. When they fall back to the lower energy level, they release the excess energy as light of a specific wavelength and color. Different metal ions have different energy levels and emit different colors of light.
Sources
- Ole Pederson. “Pharmaceutical Chemical Analysis”. (CRC press Taylor and Francis group. 2006)
- Flame Tests – Chemistry LibreTexts
Sulfate
Each general chapter (EP (2.3.1), USP <191> and JP (1.09>) presents several reactions for the identification of sulfate. The first reaction is similar (but not exactly the same) in the 3 pharmacopoeias. The 3 reactions in USP and JP are similar. The reactions are:
- Precipitation with barium chloride (present in the 3 pharmacopeia)
- Reaction with Iodine (2nd reaction in the EP)
- Precipitation with lead acetate (2nd reaction as per USP and JP)
- Addition of the hydrochloric acid (3rd reaction as per USP and JP)
Precipitation with barium chloride
This test is the 1st test in EP, JP and USP, similar (but not exactly the same) in the 3 pharmacopoeias.
Procedure
- As per EP: to an aqueous solution of sulfate are added diluted hydrochloric acid and a barium chloride solution. A white precipitate is formed.
- As per USP: to an aqueous solution of sulate is added a barium chloride solution: a white precipitate is formed that is insoluble in hydrochloric acid and nitric acid.
- As per JP: to an aqueous solution of sulate is added a barium chloride solution: a white precipitate is formed that is insoluble in diluted nitric acid.
Science behind
Most sulfate salts are soluble in water, one of the exceptions is barium sulfate (BaSO4). The identification test of sulfates (SO42-) is based in the insolubility of barium sulfate by the addition of barium chloride (BaCl2):
Ba2+ + SO42– → BaSO4↓
If carbonates (CO32-) are present, it will also precipitate barium carbonate (BaCO3), practically insoluble in water, but soluble in weak acids:
Ba2+ + CO32– → BaCO3↓
BaCO3↓ + 2HCl → Ba2+ + 2Cl– + H2O + CO2
Or
BaCO3↓ + 2HNO3 → Ba2+ + 2NO3– + H2O + CO2
Sources
- Barium Carbonate: Structure, Properties, and Uses (collegedunia.com)
- Ole Pederson. “Pharmaceutical Chemical Analysis”. (CRC press Taylor and Francis group. 2006)
Reaction with Iodine
Reaction B as per EP, used when it is suspected other species (sulfites, dithionites, iodates, selenates and tungstenates) are in the sample that give a false positive.
Procedure
- To the suspension obtained in the precipitation with barium chloride, a solution of iodine is added. The suspension remains yellow (distinction from sulfites, SO32-, and dithionites).
- Solution is decolorised by dropwise addition of a stannous chloride solution (distinction from iodates).
- Boil the mixture: no coloured precipitate is formed (distinction from selenates and tungstates).
Science behind
If the precipitate with barium chloride was due to sulfite (SO₃2-) or dithionate (S2O42-), they will reduce the iodine (I2) to iodide (I–), eliminating the yellow colour:
SO32- + I2 + H2O → SO42- + 2H+ + 2I–
S2O42- + 3I2 + 4H2O → 2HSO4 – + 6H++ 6I–
The addition of stannous chloride (SnCl2) in acidic medium will decolorize the solution by reducing the iodine (I2) to iodide (I–):
Sn2+ + 2H+ + I2 → Sn4+ + 2I–
However, if the precipitate is barium iodate (Ba(IO3)2), the iodate is reduced to iodine (I2):
2IO3– + 5Sn2+ + 12H+ → I2 + 5Sn4+ + 6H2O
and the solution will not decolorise.
About the distinction from selenates and tungstates (no coloured precipitation formed with boiling), I have not found any explanation for this. Ole Pederson (2006), source 1, says “Had the white precipitate been barium selenates or barium tungstates, they would have been reduced to elemental selenium and wolfram blue, respectively.”
Sources
- Ole Pederson. “Pharmaceutical Chemical Analysis”. (CRC press Taylor and Francis group. 2006)
- Iodometry – Wikipedia
- James P. Danehy and Charles William Zubritsky (1974) “lodometric Method for the Determination of Dithionite, Bisulfite, and Thiosulfate in the Presence of Each Other and Its Use in Following the Decomposition of Aqueous Solutions of Sodium Dithionite”, Analytical chemistry, VOL. 46, NO.3. 391-395.
- Iodine clock reaction – Wikipedia
Precipitation with lead acetate
This is the 2nd reaction in the JP and USP
Procedure
- Neutral solutions of sulfates yield and white precipitate with the addition of a lead (II) acetate solution.
- The precipitate dissolves when ammonium acetate solution is added.
Science behind
Lead (II) acetate Pb(C2H3O2)2 in presence of sulfates produced a white precipitate of lead sulfate (PbSO4):
SO42- + Pb(C2H3O2)2 → PbSO4↓ + 2 C2H3O2–
As lead(II) sulfate can be dissolved in concentrated nitric acid (HNO3), hydrochloric acid (HCl) and sulfuric acid (H2SO4) (producing acidic salts or complex compounds) and in concentrated alkali (giving soluble tetrahydroxidoplumbate(II) [Pb(OH)4]2− complexes), neutral solutions ensure that the lead sulfate is not dissolved.
The addition of ammonium acetate (CH3COONH4) produces a double displacement reaction (also called exchange reaction or metathesis reaction) [sources 2 and 3]:
PbSO4 + 2CH3COONH4 → (CH3COO)2Pb +(NH4)2SO4
However, some authors consider that this reaction is possible by the formation of complexes (CH3COO)3 Pb– in CH3COONH4 (source 4).
Sources
- Lead(II) sulfate – Wikipedia
- Lead sulphate is soluble.A.In concentrated nitric acid.B.In concentrated hydrochloric acid.C.In solution of ammonium acetate.D.In water. (vedantu.com)
- (Arthur A. Noyes and William H. Whitcomb (1905) “The solubility of lead sulphate in ammonium acetate solutions”, J. Am. Chem. Soc. 1905, 27, 6, 747–759.) (https://ia800708.us.archive.org/view_archive.php?archive=/22/items/crossref-pre-1909-scholarly-works/10.1021%252Fja01974a600.zip&file=10.1021%252Fja01984a013.pdf)
- c5ra23559f (rsc.org): “Preparation of high-purity lead oxide from spent lead paste by low temperature burnt and hydrometallurgical with ammonium acetate solution”. Cheng Ma , Yuehong Shu, Hongyu Chen. RSC advances, accepted manuscript, accessed 18 Jan 2024. Accessed 20 Jan 2024.
Addition of hydrochloric acid
This is the 3rd reaction as per USP and EP, used to distinguish sulfates from thiosulfates.
Procedure
Addtion of hydrochloric acid to a solution of sulfates (SO42-) will not form a precipitate (distinction from thiosulfates (S2O32-))
Science behind
Thiosulfates (S2O32-) in presence of hydrochloric acid (HCl) produces sulfur (S) as a cloudy yellow-white precipitate and sulfur dioxide gas (SO2) during the reaction:
S2O32- + 2HCl → 2Cl– + H2O + SO2↑ + S↓
Sources
Limit test
To do a limit test, you need to make 2 solutions: one with the substance you want to test and another with a known amount of the impurity you are looking for. Then you compare the two solutions and see which one is more clear and less coloured. The solution with the substance you want to test should be clearer and less coloured than the other one.
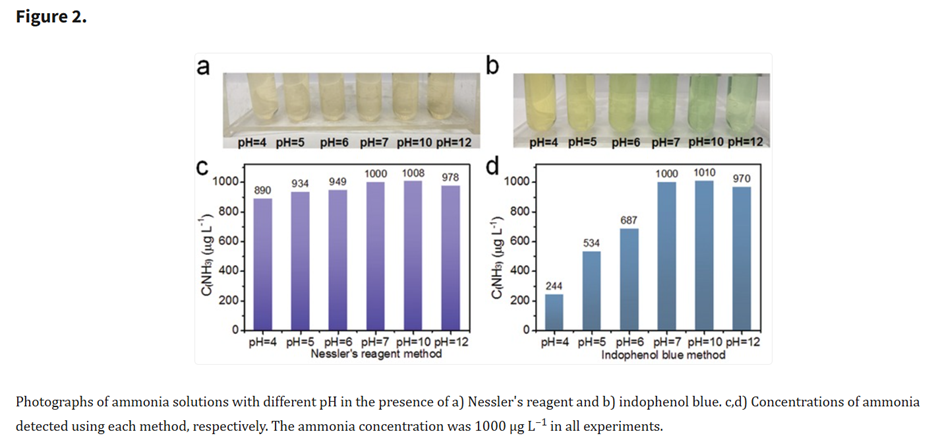
Ammonium (EP)
Ammonium as per EP (2.4.1) follow 2 methods:
- Method A: reaction with potassium tetraiodomercurate.
- Method B: reaction with magnesium oxide and silver manganese paper.
MEthod A: Reaction with potassium tetraiodomercurate.
Procedure
- Sample solution: make the sample solution alkaline if necessary and add alkaline potassium tetraiodomercurate solution.
- Standard solution (1 ppm ammonia): add alkaline potassium tetraiodomercurate solution.
- After 5 minutes, any yellow colour in the test solution is not more intense than that in the standard.
Science behind
The use of the potassium tetraiodomercurate(II) (K2[HgI4]) to detect ammonia was first proposed by Nessler in 1856. The reagent is also referred as Nessler reagent, however, the preparation varies depending on the source.
The ammonia (NH3) and the ammonium ion (NH4+) form complexes with the potassium tetraiodomercurate(II) (K2[HgI4]) of different colours depending on the concentration of ammonia (see sources 2 and 3 for some proposed reactions).
The formation of the complex is slow, which is the reason of waiting for 5 minutes to compare colours.
A pH above 7 ensures consistent readings:
Sources
- Ole Pederson. “Pharmaceutical Chemical Analysis”. (CRC press Taylor and Francis group. 2006)
- Potassium tetraiodomercurate(II) – Wikipedia
- Jing-Ping Wang, Xin-Hong Wang, Jian Chen, Zheng-Hao Fei, “Experimental exploration for measurement of ammonia nitrogen in water by Nessler’s reagent colorimetry.” Civil and Environmental Research (2019) Vol.11, No.1, pg 58 – 63) Link: Microsoft Word – CER-Vol.11 No.1 2019.docx
- Ammonia Detection Methods in Photocatalytic and Electrocatalytic Experiments: How to Improve the Reliability of NH3 Production Rates? – PMC
Method B: reaction with magnesium oxide and silver manganese paper.
Procedure
Sample solution (sample suspended or dissolved in water) and standard (1 ppm ammonia) treated in the same manner:
- In separated 25 mL jars fitted with a caps, place the sample solution and the standard.
- Add magnesium oxide.
- Close immediately after placing a piece of silver manganese paper (paper treated with a mixture of manganese sulfate and silver nitrate solutions) 5 mm square, wetted with a few drops of water R.
- Swirl, avoiding projections of liquid, and allow to stand at 40 °C for 30 min.
- Any grey colour in the silver manganese paper from the sample solution is not more intense than that of a standard.
Science behind
Magnesium oxide (MgO) is a moderately strong base with low solubility in water. When added to water, it reacts to form magnesium hydroxide (Mg(OH)₂), which only partially dissolves, making the solution weakly alkaline with a pH of around 9 to 10, depending on the conditions:
MgO + H2O → Mg(OH)2
With the addition of MgO, any ammonium in the solutions will be liberated as ammonium (NH3).
NH4+ + OH– → NH3↑ + H2O
Heating aids the evaporation of ammonium that reacts with the silver and the manganese on the paper:
4NH3 + Mn2+ + 2Ag+ + 3H2O → MnO(OH)2↓ + 2Ag↓ + 4NH4+
Sources
- Ole Pederson. “Pharmaceutical Chemical Analysis”. (CRC press Taylor and Francis group. 2006).
- acid-base behaviour of the period 3 oxides
- Magnesium oxide – Wikipedia
Arsenic
The arsenic limit test as per the general chapters of EP (2.4.2), USP <211> and JP <1.11> follow the same principles, however, there are some differences between them. This test requires special glassware, different for each pharmacopeia.
Procedure
- To the sample solution and the standard (solution with a known concentration of arsenic), are added hydrochloric acid (HCl), Tin (II) chloride (aka stannous chloride, SnCl2) and potassium iodide (KI). Wait.
- Add Zn.
- Fumes passed through lead acetate (Pb(C2H3O2)2) and collected in a solution of silver diethyldithiocarbamate.
- After a specific time both solutions are compared: the colour obtained in the test solution should not be more intense than that obtained with the standard.
Science behind
Arsenic is present as As(V) and As(III). The addition of KI reduces the As(V) to As(III). Note: I have not found reference to the specific reaction.
Once all the arsenic is converted to As(III), the addition of Zn produces arsine gas (AsH3):
As3+ + 3Zn + 3H+ → AsH3↑ + 3Zn2+
The addition of SnCl2 facilitates the arsine formation (however, I have not found further explanation). The HCl forms H2 gas that helps to sweep the arsine:
2H+ + Zn → H2↑ + Zn2+
Arsine reacts with silver diethyldithiocarbamate producing red coloured products. Coloration is compared with the colours obtained from the standard solution.
Lead acetate is used to remove hydrogen sulfide (H2S), phosphine (PH3) and stibine (SbH3). In the case of H2S, PbS (black colour) is formed:
Pb2+ + H2S → PbS↓ + 2H+
Sources
- https://www.web-formulas.com/Formulas_of_Chemistry/Limit_Test_of_Arsenic.aspx
- https://en.wikipedia.org/wiki/Silver_diethyldithiocarbamate
- Ole Pederson. “Pharmaceutical Chemical Analysis”. (CRC press Taylor and Francis group. 2006)
Bromide
This test is used in the sodium chloride monograph in EP, JP and USP. The monograph has been harmonized between the 3 pharmacopoeias.
Procedure
- Dissolve the sample in water and prepare an aqueous standard solution containing a known concentration of bromide. Both samples are treated concomitantly.
- To each sample, add a phenol red solution with a pH 4.7 and a dilute solution of chloramine.
- After 2 minutes, add diluted sodium thiosulfate and mix.
- Dilute to 10.0 mL.
- Measure the absorbance at 590 nm. The absorbance of the sample solution should not be greater than the absorbance of the standard solution.
Science behind
The reaction is based on the oxidation of the bromide (Br–) to bromine (Br2) by the chloramine, and the subsequent reaction of the bromide with the phenol red to form bromophenol blue:
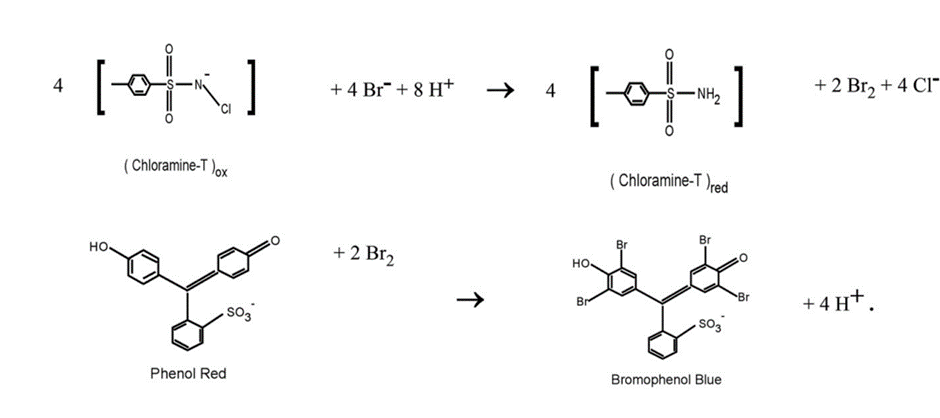
Source: Improved bromide measurements using chloramine-T shows, bromide depletion in the Gulf of Maine (sciencedirectassets.com) (John P. Christensen. Continental Shelf Research 193 (2020) 104028)
The addition of sodium thiosulfate stops the reaction by decomposition of the excess of chloramine, which would otherwise oxidize the bromophenol blue. I have not found the specific reaction, however, thiosulfate is widely used to remove chloramine, see for example, About Chloramine | Fritz Aquatics.
590 nm is in the region of maximum absorbance of the bromophenol blue:
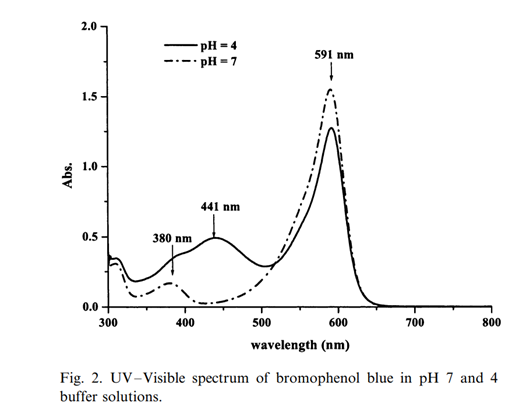
Source: (PDF) Determination of bacterial activity by use of an evanescent-wave fiber-optic sensor (researchgate.net) A. Kishen et al. Biosensors and Bioelectronics 18 (2003) 1371-1378.
Sources
- Improved bromide measurements using chloramine-T shows, bromide depletion in the Gulf of Maine (sciencedirectassets.com) (John P. Christensen. Continental Shelf Research 193 (2020) 104028).
- PII: 0039-9140(89)80057-8 (uci.edu). David R. Jones (1989) “Difficulties with the chloramine-t-phenol red method for bromide determination”. Talanta, Vol. 36, No. 12, pp. 1243-1247, 1989
- About Chloramine | Fritz Aquatics.
- (PDF) Determination of bacterial activity by use of an evanescent-wave fiber-optic sensor (researchgate.net) A. Kishen et al. Biosensors and Bioelectronics 18 (2003) 1371-1378.
Chloride
This limit test is described in the EP (2.4.4), JP <1.03> and USP <221>. The principle of the reaction is similar in all cases with some minor differences between the different pharmacopeia.
Procedure
- To the sample solution (in the USP specify that it has to be neutralized with nitric acid first).
- Add nitric acid, silver nitrate and mix.
- Prepare a standard solution with a known concentration of chlorice and treat in the same manner than the sample solution.
- After standing for 5 min protected from light, any opalescence in the test solution should be not more intense than that in the standard.
The science behind
The addition of silver nitrate (AgNO3) leads to the precipitate of silver nitrate:
Ag+ + Cl– → AgCl↓
The presence of nitric acid excludes a large number of substances. One group is the silver salts of weak acids that are unstable in dilute nitric acid and decompose to gas, such as carbonate and sulfite.
Sources
- Ole Pederson. “Pharmaceutical Chemical Analysis”. (CRC press Taylor and Francis group. 2006)
Heavy metals
While there isn’t a universally agreed-upon definition for heavy metals, they are typically recognized as metals that possess high densities, atomic weights, or atomic numbers (source 1). Heavy metals are covered by EP (2.4.8), JP <1.07>, and USP <231>. There are several methods to prepare the sample in all pharmacopoeias (8 methods in EP, 4 in JP, and 3 in USP), but they all follow the same principle: the sample reacts with a thioacetamide solution (JP) or a sulphide solution (USP and EP) to form sulfide salts. The USP and EP allow the use of a thioacetamide solution as an alternative.
Procedure
- Preparation of ‘Sample Solution’: The preparation process can vary depending on the pharmacopoeia and the characteristics of the sample. I won’t go into detail about each method for the sake of brevity.
- ‘Standard Solution’: This refers to a solution that contains a known concentration of lead.
- Additional Solution: Both the EP and USP often use an additional solution to confirm the suitability of the test. The EP uses a ‘blank’ solution, which is a diluted version of the ‘sample solution’. Conversely, the USP uses a ‘monitor’ solution, which is made by adding a known quantity of lead to a ‘sample’ solution.
- Acidification of the Solutions: All solutions are acidified. In the case of EP and USP, this is done by adding a pH 3.5 buffer, while the JP uses an acid.
- Addition of a Sulfide or Thioacetamide Solution: A sulfide solution is added in the JP method, while a thioacetamide solution is used in the EP and USP methods. However, both EP and USP also allow the use of a sulfide TS solution as an alternative.
- Comparison of the Colours: After waiting for a few minutes, the colours of the solutions are compared. The colour of the sample solution should not be more intense than that of the standard solution. If a third solution is required by the method, a suitability test would be conducted (as in the EP and USP methods).
Science behind
The science behind of the test is based on the highly insolubility of sulfides that can be clasisfied as heavy metals: lead, bismuth, mercury, … and involves the formation of a colloidal precipitate of these insoluble sulfide salts. This occurs when aqueous solutions of these metal cations interact with sulfide sources (H2S, NaHS, Na2S) , resulting in the precipitation of solid sulfides. For instance, the reaction between hydrogen sulfide (H2S) and lead (Pb) yields lead sulfide (PbS):
Pb2+ + H2S → PbS↓ + 2H+
The pharmacopoeia utilizes either a solution of sulfides (made from sodium sulfide: Na2S) or a solution of thioacetamide as sulfide sources. In the case of thioacetamide, the solution is prepared with sodium hydroxide (NaOH), and the thioacetamide (CH3-CS-NH2) undergoes hydrolysis:
CH3-CS-NH2 + H2O → CH3-COO– + NH4+ + H2S
The pH level of the solution plays a crucial role in the test’s selectivity. Variations in pH are employed to separate different cations through selective precipitation and to identify unknown cations via group separation.
Sources
- Heavy metals – Wikipedia
- The_Solubility_Rules.pdf (tamu.edu)
- Sulfide Ion (S²⁻) – Chemistry LibreTexts
- Sulfide – Wikipedia
- Ole Pederson. “Pharmaceutical Chemical Analysis”. (CRC press Taylor and Francis group. 2006)
Iron as per EP
The limit test of iron as per EP is given in the general chapter (2.4.9). In some monographs (like the arginine hydrochloride) there is an initial step to extract the iron from the sample with the aid of methyl isobutyl ketone (MIBK, also known as 4-methyl-2-pentanone). I cover the separation with MBIK and the general chapter here.
Extraction of iron with methyl isobutyl ketone (MIBK)
Procedure
- In a separating funnel, dissolve the sample in diluted HCl.
- Add the MBIK, shake and separate the aqueous phase (dilute HCl) and the organic phase (MBIK). This step is typically done 3 times, shaking the sample in diluted HCl with 3 portions of MBIK. At the end, the different portions of MBIK are combined and the original aqueous solution (sample in diluted HCl) is discarded. NOTE: from the literature review, timings and number of separations are critical.
- Add water to the combined portions of MBIK, shake and separate the aqueous phase. This aqueous phase is used to perform the limit test as per general chapter (2.4.9)
Science behind
The procedure used in the separation phase is widely used in extractive metallurgy (see references 1 and 2 below), however, I have not found a detailed mechanism of the science behind it. Based on a publication from the Royal Society of Chemistry (reference 3), which gives a good general review on the topic, my understanding of the science behind is:
- In acidic conditions, the iron form a water insoluble complex with MIBK, that is retained by the MIBK. I have not found a reliable source of the complex formed.
- When the pH is increased, the iron complex insoluble in water is no longer stable and the iron is dissolved in water.
The change of pH is widely used to extract metals in the metallurgy, as shown in the figure from reference 3
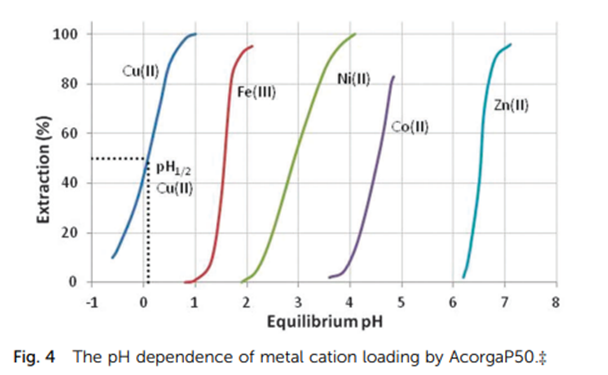
References
- Farouq, Selim (2017), J. Chil. Chem. Soc. Solvent extraction of iron ions from hydrochloric acid solutions, 62, Nº 2
- Want et al (2012). Hydrometallurgy. Simultaneous oxidation and extraction of iron from simulated ilmenite hydrochloric acid leachate, 129-130, 105-110
- Wilson et al, 2014, Chem Soc. Rev ‘Solvent extraction: the coordination chemistry behind extractive metallurgy’, 43, 123.
Limit test as per EP (2.4.9)
Procedure
- To the solution containing the sample and to a solution containing the required standard iron solution, add citric acid, thioglycolic acid and mix.
- Add ammonia until alkaline reaction.
- Dilute both solutions to 20 mL with water.
- After 5 min, any pink colour in the test solution is not more intense than that in the standard.
Science behind
- Citric acid prevents the precipitation of iron hydroxide by ammonia and also forms a soluble complex with other metal ions that might interfere with the test.
- Liliana Gonzalves (liliana.goncalves1@mdlz.com) (Thanks Lilina!!!) has provided with the following details: Thioglycolic acid (HS-CH2-COOH) reduces any iron (III) to iron (II) and then reacts with it to form ferrous thioglycolic, which undergoes Ligand-Metal charge transfer that has the energy gap of this transition matches a wavelength outside of the visible absorption spectrum. This is however changed with the deprotonation of the carboxylic acid (lowest pka) which shifts the light absorption of the complex resulting in the solution producing a red colour.
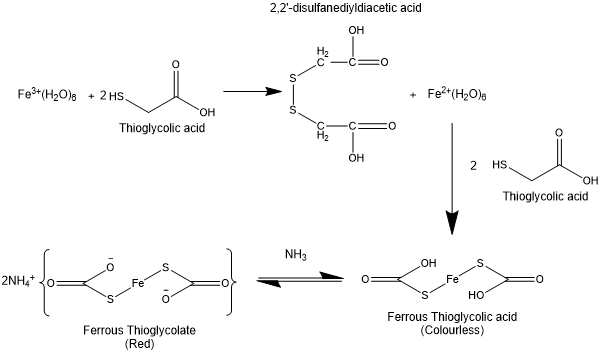
References
- Limit Test for Iron – Principle, Apparatus, Procedure and notes (learnaboutpharma.com)
- Ole Pederson. “Pharmaceutical Chemical Analysis”. (CRC press Taylor and Francis group. 2006)
- Rhaman et al. (2020), ‘Method validation on iron determination by spectrophotometric method in aqueous medium’, Indian Journal of Chemistry, Vol. 59A, pp. 790-796.
Iron as per JP <1.10> Method a
Thanks to Liliana Goncalves (Liliana.Goncalves1@mdlz.com) for helping with this test.
Procedure
- To the test solution and the control solution add dilute L-ascorbic acid (1 in 100), mix well, and allow to stand for 30 minutes.
- Add a, a’-dipyridyl (also known as 2,2 bipyridil) in ethanol, water and allow to stand for 30 minutes.
- Compare the colours developed in both solutions against a white background. The test solution has no more colour than the control solution.
Science behind
L-ascorbic forms complexes with iron (III) and reduces it to iron (II) (source 1). It also adjust the pH to prevent the precipitation of Fe(OH)2 and/or Fe(OH)3.
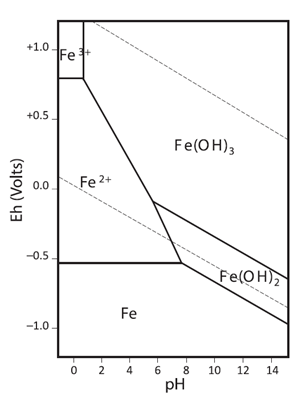
Pourbaix diagram of iron. Source: John Monhemius (2017) “The iron elephant: A brief history of hydrometallurgists’ struggles with element no. 26”, CIM Journal 8(4): 197-206.
The iron (II) form complexes with a, a’-dipyridyl:
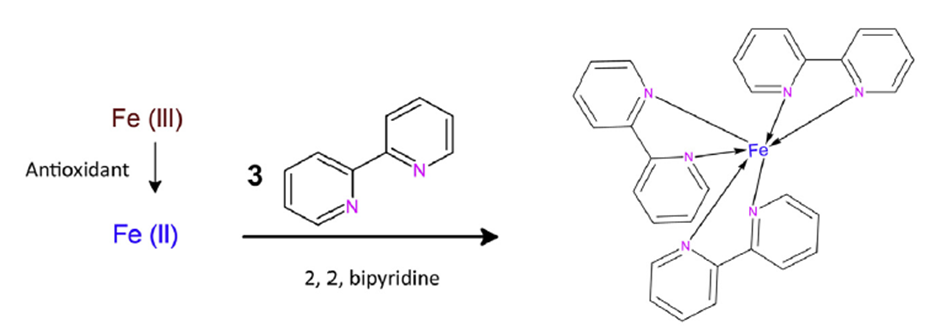
2,2-bipyridine iron (II) complex. Source: Ferric-bipyridine assay: A novel spectrophotometric method for measurement of antioxidant capacity: Heliyon (cell.com).
The complex is chiral, giving 2 isomers:
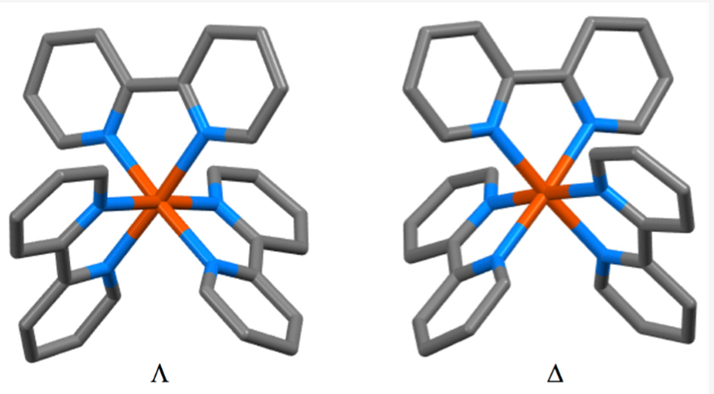
2,2-bipyridine iron (II) complex isomers. Source: Molecules | Free Full-Text | The Early Years of 2,2′-Bipyridine—A Ligand in Its Own Lifetime (mdpi.com)
Sources
- Ascorbate oxidation by iron, copper and reactive oxygen species: review, model development, and derivation of key rate constants | Scientific Reports (nature.com).
- Ferric-bipyridine assay: A novel spectrophotometric method for measurement of antioxidant capacity: Heliyon (cell.com).
- Molecules | Free Full-Text | The Early Years of 2,2′-Bipyridine—A Ligand in Its Own Lifetime (mdpi.com)
Iron as per USP <241>
Procedure
- Two aqueous solutions are prepared: one with the sample and another with a known concertation of iron (standard). Hydrochloric acid is added during the preparation of these solutions.
- Ammonium peroxydisulfate and ammonium thiocyanate solution are added to both solutions.
- The colours are compared, the colour of the sample solution should be not darker than the colour of the standard solution.
Science behind
The addition of hydrochloric acid reduces the pH to prevent the formation and precipitation of iron hydroxide, Fe(OH)3 and/or Fe(OH)2.
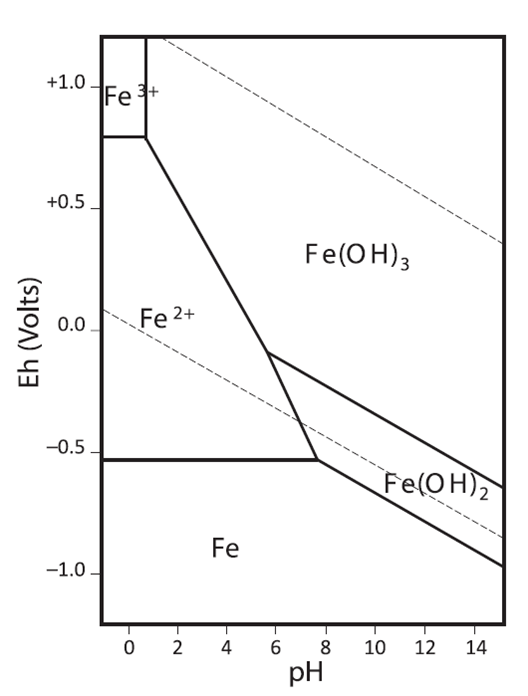
Pourbaix diagram of iron. Source: John Monhemius (2017) “The iron elephant: A brief history of hydrometallurgists’ struggles with element no. 26”, CIM Journal 8(4): 197-206.
The peroxydisulfate (S2O82-) oxidises the iron (II) to iron (III):
S2O82- + 2Fe2+ → 2SO42- + 2Fe3+
The iron (III) with thiocyanate (SCN–) forms the complex iron(III) thiocyanate that has a deep red colour:
[Fe(H2O)6]3+ + SCN– → [Fe(SCN) (H2O)5]3+
Sources
- Chemistry of Iron – Chemistry LibreTexts
- Catalysis (6.2.7) | AQA A Level Chemistry Revision Notes 2017 | Save My Exams
- FERRIC THIOCYANATE (ncats.io)
- Want et al (2012). Hydrometallurgy. Simultaneous oxidation and extraction of iron from simulated ilmenite hydrochloric acid leachate, 129-130, 105-110
Phosphate
Procedure as per EP (2.4.11)
Procedure
- Mix the sample solution with Sulfomolybdic reagent (aqueous solution of ammonium molybdate with sulfuric acid) and shake.
- Add an aqueous solution of stannous chloride with hydrochloric acid.
- Prepare a standard solution of known concentration of phosphate and treat on the same manner.
- After 10 minutes compare the colours of both solutions. Any colour in the sample solution should not be more intense than that in the standard.
Science behind
The reaction of the phosphates (PO43-) with the ammonium molybdate ((NH4)6Mo7O24, however, for simplicity, in this reaction, it is usually written as MoO42–) produces yellow ammonium phosphomolybdate ((NH4)3[P(Mo3O10)4) sometimes written as (NH4)3PMo12O40):
HPO42– + 3NH4+ + 12MoO42– + 23H+ → (NH4)3[P(Mo3O10)4] + 12H2O.
In the second step, the reductant stannous chloride (SnCl2) reduces the yellow ammonium phosphomolybdate to a substance called ‘molybdenum blue’. “Several possible ‘molybdenum blue’ species are identified as end products of the reaction depending on the conditions used.” (source 2).
Sources
- Ole Pederson. “Pharmaceutical Chemical Analysis”. (CRC press Taylor and Francis group. 2006)
- Edward A. Nagul, Ian D. McKelvie, Paul Worsfold and Spas D. Kolev: “The molybdenum blue reaction for the determination of orthophosphate revisited: Opening the black box”. Analytica Chimica Acta, Volume 890, 26 August 2015, Pages 60-82.
Sulfate
Procedure
JP <1.14> and USP <221>:
- The sample and the standard solutions are acidified and barium chloride (BaCl2) added and mixed.
- After several minutes, the turbidity of the solutions are compared. Any turbidity in the test solution is not more intense than that in the standard.
EP (2.4.13) follow the same procedure, but it adds a solution of BaCl2 mixed with sulfate (SO42-) in 30% ethanol before acidifying the sample.
Science behind
Most sulfate salts are soluble in water, one of the exceptions is barium sulfate (BaSO4). The limit test of sulfates (SO42-) is based in the insolubility of barium sulfate by the addition of barrium chloride (BaCl2):
Ba2+ + SO42– → BaSO4↓
The role of the mixture an excess of BaCl2 with SO42- in 30% ethanol is to prepare a crystal seeds, giving a precipitate of well-defined particle size distribution for seeding crystals.
Acidic conditions (even mildly acidic) are used to dissolve barium salts that precipitate in neutral conditions (carbonate, sulfite, and phosphate).
Sources
- Ole Pederson. “Pharmaceutical Chemical Analysis”. (CRC press Taylor and Francis group. 2006)
- Zimmermann, J., Krogh-Svendsen, E., and Reimers, F., Limit tests for impurities IV. Investigations into the reproducibility of precipitation part III, Analytica Chim. Acta, 16, 1957, 6.